罕见神经发育遗传病——天使综合征
转自知乎:https://zhuanlan.zhihu.com/p/34418193.
作者:谷雨
一、什么是罕见神经发育遗传病
大约有2~3%的儿童有智力障碍(intecllectual disability),而70%的智力障碍是由基因变异导致的。目前,已知有600多种基因能成为智力障碍的诱因。
1985年,人们发现了第一个神经发育的致病基因。此后,智力障碍才得以被诊断出来。2000年,由于人类基因组计划的开展,越来越多的神经发育致病基因被发现,从而引领了神经发育障碍诊断的第二次飞跃。
对神经发育的研究也许无法治愈智力障碍儿童,但是可以大幅改善其生活质量。这也是进行神经发育研究的意义之一。
下图列出了目前已知的神经发育障碍致病基因,他们大多数与RAS/ERK/mTOR分子通路有关。其中,NF1基因变异最有可能导致神经发育障碍,病情也最严重。两个原因:
1)NF1基因表达的是NF1蛋白,即神经纤维素I(Neurofibromin I)。从图中我们可以看到,它处于许多分子机制的上游,因此一旦变异,就会使下游的许多过程发生变化;
2)许多影响神经发育的蛋白都是兴奋性的,但NF1蛋白为抑制性蛋白。当它无法起到正常起作用时,其调控的下游通路会出现功能增益(gain of function),过度活跃,过度合成蛋白。
合成的蛋白增多很可能就会出现如蛋白质聚集等扰乱细胞正常工作的错误。如果只是其中一个兴奋性蛋白出了问题,虽然可能导致功能减弱(loss of function),但很有可能有其他功能相同或相似的蛋白能够起到补偿作用。
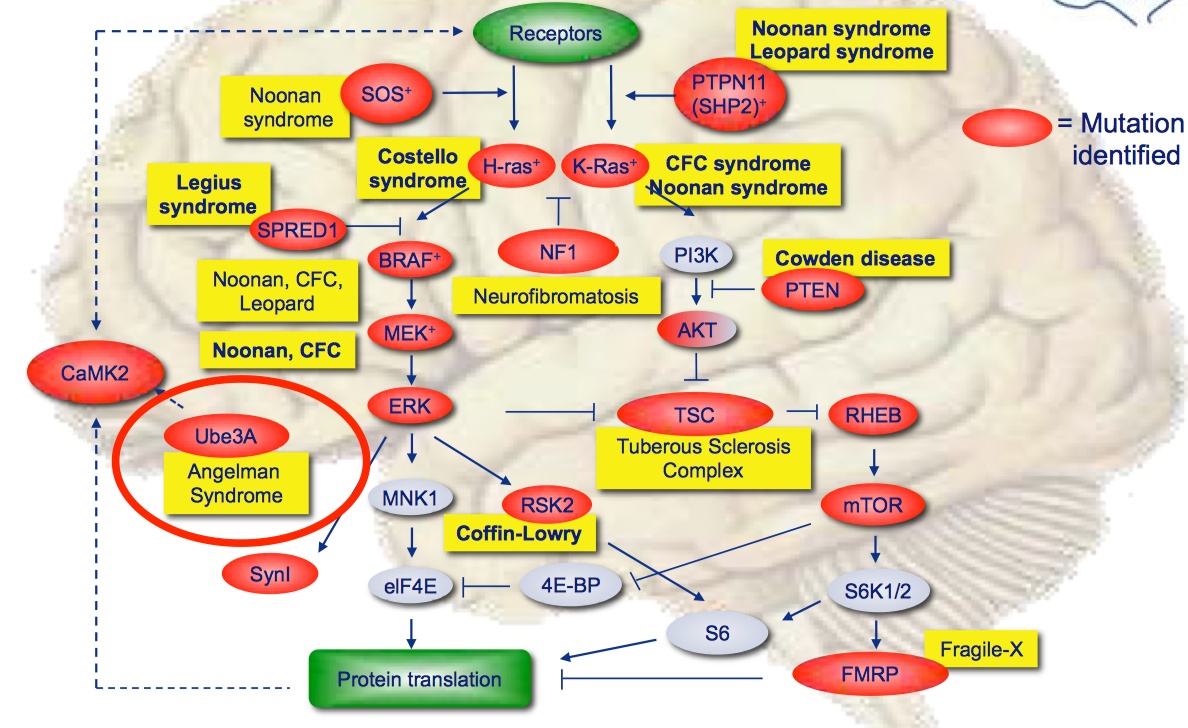
CaMKII变异对神经发育障碍的影响是直到去年才被检测出来的(Küry et al., 2017)。明明我们在二三十年前就知道了它的存在,为什么现在才发现它的变异导致的致病影响呢?这是因为许多其他基因的变异会发癌变,使患者表现出明显的躯体症状,从而容易对患者进行识别和归类。对多个相同症状的患者进行基因检测,就容易得知他们的共同致病基因。这也是为什么许多神经发育障碍的名字中都有“综合征”字样,如天使综合征等。但是CaMKII的病变不如其他明显,难以将该类病变人群归类,因而难以检测致病基因。
二、CaMKII对疾病的影响
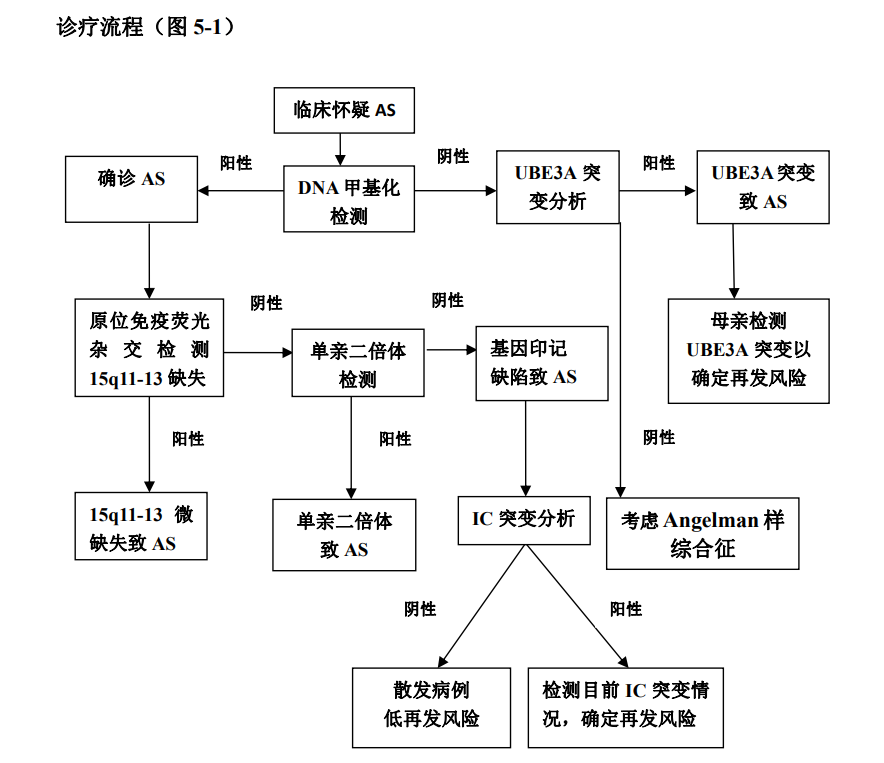
1. 天使综合征(Angelman Syndrome,AS)
AS的发病率为两万分之一。患者表现出智力障碍、言语缺失、癫痫及EEG异常、运动缺陷、睡眠缺陷和行为异常(如自闭症谱系障碍[Autism spectrum disorder, ASD]、进食障碍、睡眠障碍等)。不过,患者们总是常常面露笑容,有天使般的神态。巧合的是,首先发现该疾病,并系统描述它的医生Harry Angelman,其名字里也有“天使“一词。
AS是由神经系统内母本15号染色体上UBE3A基因功能缺陷无法正常表达而导致的。
UBE3A蛋白(Ubiquitin-protein ligase 3A)就是E6AP蛋白,是泛素蛋白连接酶3型的一种。泛素-蛋白溶酶体是生物体内降解蛋白质的两种途径之一,另一种是自噬-溶酶体途径。降解蛋白质与细胞周期、免疫应答、代谢物清理等有关。简单来说,泛素-蛋白溶酶体途径是通过3种酶活化泛素,并将其连接到被降解的蛋白质上做好标记。这样,被标记好的蛋白质就会被送回工厂分解回收。其中的一种酶,泛素蛋白连接酶3型,就负责将活化的泛素连接到即将被降解的蛋白质上。当然了,蛋白质种类繁多,你很容易就猜到,这个连接酶一定是有特异性识别功能的。UBE3A是众多连接酶中的一种,但目前,其关键靶向未知。它最初是在关于人类乳头瘤病毒(HPV-16)发病机制的研究中被发现的,那个时候的名字是E6AP(E6-associated protein),故事相当有趣,感兴趣的可以去查阅相关内容。
目前,研究者们通过基因打靶(gene targeting)敲除母本UBE3A基因后获得了天使综合征的小鼠模型。这种小鼠模型除了无法表现出言语缺失外完全模拟了人类天使综合征。
2. CaMKII对天使综合征的影响
研究发现,降低AS小鼠中增高的CaMKII T305位点上的抑制性磷酸化能恢复AS表型(Weeber et al., 2003; Van Woerden et al., 2007)。也就是说,CaMKII的活性在天使综合征中被抑制了。那么是因为这个才导致了神经疾病吗?
一个简单的想法是,让我们移除AS小鼠中的CaMKII的刹车吧。看看提高CaMKII活性会发生什么!
研究者让携带UBE3A基因的AS小鼠与αCaMKII-TT305/6VA小鼠(αCaMKII T305位点无法进行抑制性磷酸化)杂交,双方均为杂合小鼠。这样会产生四种基因型的小鼠:
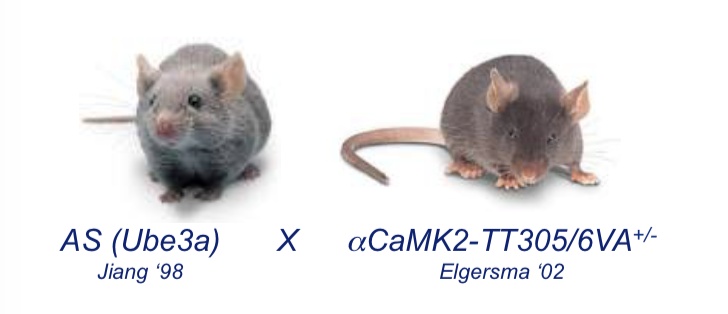
- 健康的野生型小鼠(WT);
- 健康的只携带一个αCaMKII变异基因的小鼠(αCaMKII);
- 病态的只携带父本UBE3A的AS小鼠(AS);
- 携带父本UBE3A及父本αCaMKII变异基因的小鼠(AS/αCaMKII)。
AS小鼠会表现出肥胖、癫痫、运动协调障碍和认知缺陷。而:
- AS/αCaMKII小鼠没有出现肥胖;
- AS/αCaMKII小鼠的癫痫发作显著减少;
- AS/αCaMKII小鼠在衡量运动协调的平衡杆和转动杆任务中表现明显比AS小鼠好,接近野生控制组水平。令人意外的是,αCaMKII变异小鼠甚至还在运动协调能力上表现出大幅提升;

- 在用以测试空间学习能力的Morris水迷宫实验中,AS/αCaMKII小鼠比AS小鼠好,与野生控制组无差异;
- 在恐惧学习(环境条件化)实验中,AS小鼠学习认识危险环境更慢,但AS/αCaMKII小鼠和野生控制组表现出相同水平的学习能力;
- AS/αCaMKII小鼠和αCaMKII小鼠的LTP水平均与野生控制组无差异,而AS小鼠则没有LTP;
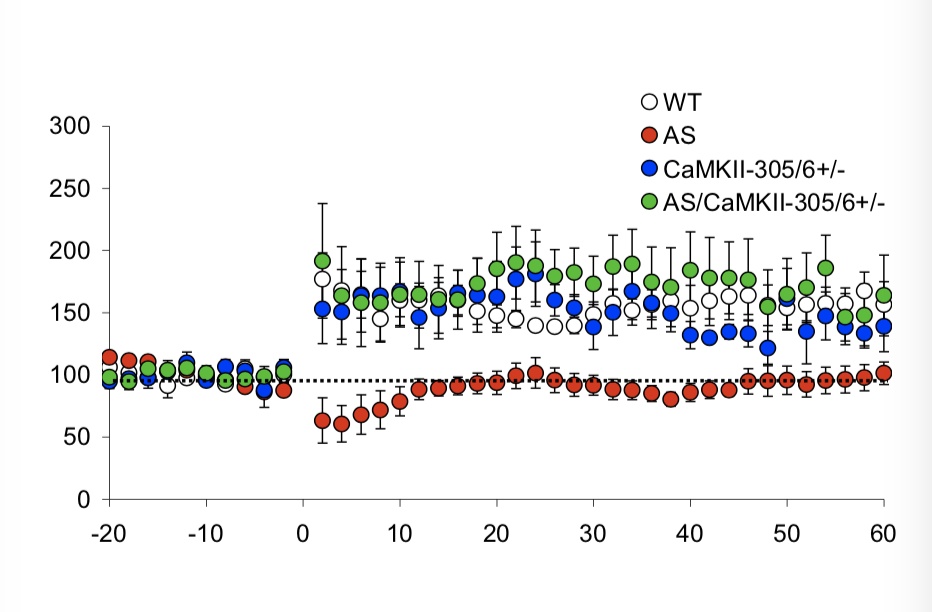
因此,我们可以下结论说,AS/αCaMKII双重变异拯救了AS单一变异的CaMKII活动性、LTP缺陷、肥胖症状、癫痫症状、认知缺陷和运动协调缺陷。
既然如此,那AS是一种病症可逆的发育性综合征吗?
天使综合征目前的治疗方案研究主要走以下两条路:
- 调节下游靶位或效应器(如CaMKII)的药物
- 激活父本UBE3A基因的药物
3. 基因印刻(gene imprinting)与天使综合征
为什么要激活父本UBE3A基因?
我们之前提到,AS是一种母本UBE3A基因无法正常表达的疾病。我们知道,每一对染色体都包含一条母本拷贝和一条父本拷贝,但它们的贡献在某些情况下不是对等的。在除神经系统外的其他部位,母本和父本染色体上的UBE3A基因都能正常表达,而在神经系统中父本基因拷贝被组织特异性沉默了,也就是被印刻了。
可能有人看到印刻就会想起“小鸭认妈”的中学生物学教材的典例,楼主表示,从心理学跑过来,在课上第一次听到“gene imprinting”,以为基因都会认妈,而旁边同学们还点头表示很懂,于是恍恍惚惚惊到怀疑世界,手机查了好几遍imprinting这个词。但是吧,它的意思勉勉强强也差不多,就是说这个基因一开始就被封印了,不许说话。
1997~1998年的一系列遗传学研究表明,神经系统过多表达UBE3A会导致自闭症。这大概就是为什么神经系统沉默了其中的一个基因。
沉默基因的方法有好些,其中最普遍的方式是甲基化修饰其DNA区段。但UBE3A的印刻并不是这样的。它很特殊。Beaudet实验室在2015年发现,与父本UBE3A基因反向的一个位点会表达一个非编码RNA(UBE3A-ATS,不编码蛋白质),这个RNA会一直产生覆盖UBE3A基因的部分,导致它无法正常表达。这个过程叫反义转录(anti-sense transcript)。这名字也是很有趣了,anti-sense之后这基因果然无法make sense。
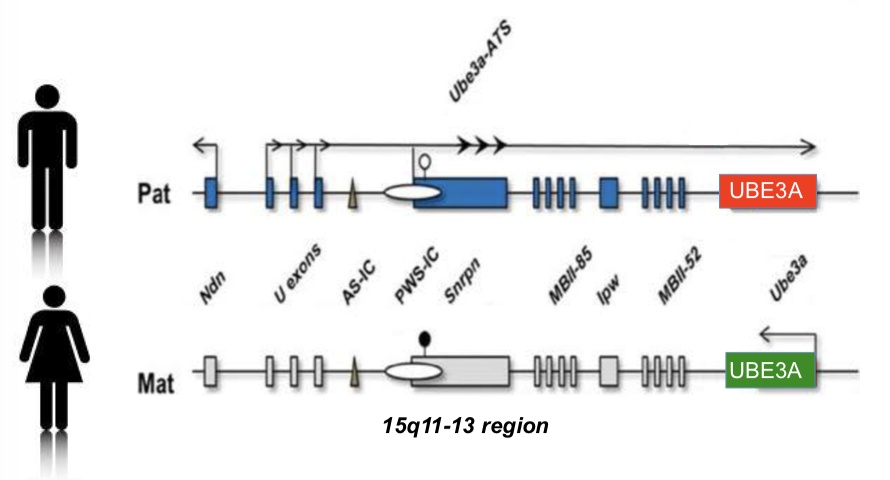
所以就有人说,那我们去掉这个反义转录,解除封印,让父本UBE3A正常表达,不就可以弥补母本UBE3A的表达缺陷了吗?
确实,黄宪松博士用拓扑异构酶抑制剂Topotecan确实能有效打开沉默的父本UBE3A,缓解AS症状,但是这种药物的毒性较大,目前仍需改善,另外,反义寡核苷酸(antisense oligonucleotide, AON/ASO)也能调节降解UBE3A的反义转录,且基本上能完全恢复UBE3A的表达(相关研究文章尚未公开发表)。
另一个问题是,AS表型的可逆性是否存在一个关键期?什么时候用药最有效?要怎么研究?
4. AS表型可逆性的关键期
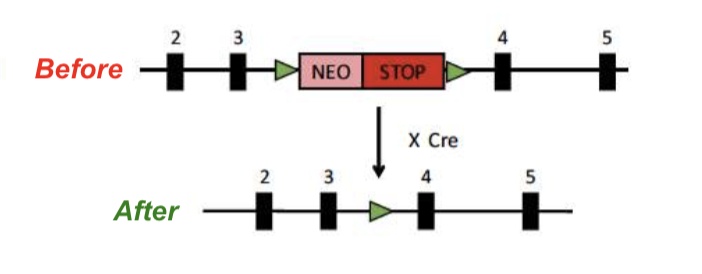
研究者创造了一种Tamoxifen-Cre AS小鼠。在正常小鼠的母本UBE3A基因中用Cre-LoxP方法插入一个带标记停止子后,UBE3A无法正常表达,此时小鼠就是AS表型的小鼠;若给予药物Tamoxifen,则能启动Cre重组酶,剪掉停止子,让UBE3A基因正常表达。如此一来,在不同时期给予Tamoxifen,就能观测到不同时期AS表型的可逆性了!
结果发现,在任何年龄(幼年、青少年期和成年期)恢复UBE3A表达都能拯救海马LTP缺陷;前额叶皮质第5层锥体神经元的突触传递缺陷可在成年期恢复UBE3A表达时被拯救;运动缺陷只能在幼年时期恢复基因表达来拯救;筑巢行为缺陷在小鼠三周龄后恢复基因表达而被拯救。简单说,确实越早越可逆,救比不救要好。
那么,UBE3A在神经元发育完成后还有作用吗?
检验的操作时,用Tamoxifen-Cre、LoxP夹心UBE3A基因替换野生型母本基因,这样在不同年龄时期给予Tamoxifen就可以剪掉整个UBE3A基因,使它从正常小鼠变成AS小鼠。
结果是,早期发育时敲除基因会导致AS表型,3周龄后敲除只会影响到突触可塑性、筑巢行为和强迫游泳表现,12周龄后敲除仅影响突触可塑性。
至此,我们可以得出有关治疗方案和窗口期的结论:
- 用ASO可以恢复UBE3A基因表达
- 临床试验需考虑到某些待拯救行为方面的发育窗口期
- 绝大多数AS小鼠的表型在出生后的发育期内表现出来,但UBE3A蛋白对成年鼠的正常突触可塑性来说也是必不可少的。